The decade that rewrote the rules
In 2016, a landmark paper in Value in Health laid out a clear diagnosis: decisions affecting patients were being made on expert judgment alone. The clinical evidence existed. The methodology to systematically incorporate patient perspectives did not.
The EMA's Benefit-Risk Methodology Project, the IMI-PROTECT consortium, and pilot studies across Europe were generating promising tools, discrete choice experiments, multicriteria decision analysis, preference elicitation. But no single method was enough. And making any of it work required collaboration across regulators, HTA bodies, patient organisations, and industry at a scale that didn't exist yet.
It would take a decade.
What is CIOMS WG XII and why does it matter?
In May 2025, CIOMS published the report of Working Group XII: Benefit-Risk Balance for Medicinal Products. It supersedes the WG IV framework from 1998, a 27-year replacement cycle that tells you everything about how slowly this field has moved, and how significant this moment is.
The connection to the 2016 paper is direct, not coincidental. One of the lead IMI-PROTECT researchers cited in it, Shahrul Mt-Isa, went on to be a principal contributor to CIOMS WG XII.
Here's what changed:
1. The patient voice is structural, not decorative
Patient-centric benefit-risk endpoints must be designed into clinical trials from Phase I, not layered on after submission as a qualitative footnote. Patient preference studies, patient-reported outcomes (PROs), and quality-of-life data are no longer optional inputs. They are foundational.
2. BRA is continuous, not episodic
CIOMS WG XII introduces the Benefit-Risk Assessment Document (BRAD): a living record updated at every milestone from pre-clinical through post-approval. One document connecting IND submissions, Investigator Brochures, Risk Management Plans (RMPs), PBRERs, and CTD modules. The days of rebuilding the benefit-risk narrative from scratch at each regulatory touchpoint are, by design, over.
3. Uncertainty gets a seat at the table
No more acknowledging uncertainty in a footnote and moving past it. CIOMS WG XII integrates uncertainty management directly into the structured BRA framework, across study design, enrolled populations, data completeness, and statistical methods.
4. Quantitative methods: use them when they matter
Additional quantitative analysis is warranted when the trade-off is marginal and fit-for-purpose data exists. Not always. Not never. This matches the 2016 conclusion: the method must fit the problem.
The real challenge: frameworks don't ship
Understanding this evolution matters. But frameworks don't execute themselves and that's where the conversation gets uncomfortable.
This shift was also not exclusively European. The FDA's Structured Benefit-Risk Framework (2013, PDUFA V) drove a parallel move toward systematic, transparent BRA in US drug review. The direction is global. The operational readiness is not.
Three problems from 2016 remain unsolved:
The patient evidence gap. Patient advocates in CHMP meetings need validated data, not anecdote. Generating that data through preference studies, PRO instruments, and quality-of-life measures requires planning from early development. Most teams start too late.
The living document problem. A BRAD that actually gets updated at every milestone means benefit-risk evidence must flow across IND submissions, Investigator Brochures, RMPs, PBRERs, and CTD modules without being rebuilt each time. That's an infrastructure challenge, not a methodology one.
The complexity ceiling. Multiple benefits, multiple risks, uncertainty across subgroups, expanding indications, novel mechanisms. A decade on, this has only gotten harder, specially for rare disease and combination therapies.
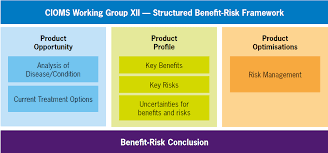
What ArcaScience was built for
The frameworks have arrived. The methodology has matured. What the field still lacks is the operational infrastructure to make it real.
ArcaScience closes that gap. The platform structures benefit-risk evidence across the full product lifecycle, synthesizes it in formats regulators and HTA bodies can act on, and maintains the traceability that turns a BRA from a periodic exercise into a defensible, living decision record.
Good intentions are not the bottleneck. Operational capacity is.